有機金属化学において、移動挿入反応は、金属錯体上の2つの配位子が結合する反応の一種です。これは挿入反応に非常によく似た反応のサブセットであり、両者は生成物の立体化学をもたらす機構によって区別されます。しかし、機構が不明な場合があるため、両者はしばしば互換的に使用されます。したがって、移動挿入反応、または略して挿入反応は、機構ではなく、1つの化学物質が通常は2番目の化学物質の既存の結合に挿入される全体的な位置化学によって定義されます。例:[1]

概要
移動挿入反応では、陰イオン(X)配位子と中性配位子が反応し、新たな陰イオン配位子を生成します。反応する陰イオン配位子と中性配位子は隣接しています。前駆体錯体が配位飽和している場合、移動挿入反応はしばしば配位不飽和生成物をもたらします。その後、新たな(中性)配位子が金属と反応し、さらなる挿入反応へとつながります。この反応は、オレフィン重合のように、単一の金属上で何度も起こることがあります。
陰イオン性配位子は、H −(ヒドリド)、R −(アルキル)、アシル、Ar −(アリール)、またはOR −(アルコキシド)です。これらの基の移動能力は、移動適性と呼ばれます。中性配位子は、CO、アルケン、アルキン、場合によってはカルベンです。
移動挿入には多様な反応が考えられます。一つのメカニズムは、陰イオン性配位子が中性配位子の求電子部位を攻撃する(陰イオン性配位子が中性配位子へ移動する)というものです。もう一つのメカニズムは、中性配位子が金属と陰イオン性配位子の間に挿入されるというものです。
CO挿入
一酸化炭素を金属-炭素結合に挿入してアシル基を形成する反応は、カルボニル化反応の基礎であり、多くの商業的に有用な生成物を生み出します。反応機構:研究により、アルキル基が分子内を移動することが明らかになっています。[2] [3]

八面体錯体のCO挿入反応経路

初期の研究では、 CH 3 Mn(CO) 5をアセチル誘導体に変換する方法が研究されました。 [4] 13 COを使用すると、生成物はシス[Mn(COCH 3 )( 13 CO)(CO) 4 ]です(図1)。

CO挿入は必ずしも転位を伴うわけではない。CpFe(L)(CO)CH 3を13 COで処理すると、アルキル転位生成物と、結合カルボニル基がメチル基に真に挿入された生成物の両方が混合して得られる。生成物の分布は溶媒の選択によって影響を受ける。[5]
平面四角形錯体のアルキル誘導体は、特に容易にCO挿入反応を起こす。平面四角形錯体における挿入反応は、その産業応用の観点から特に興味深い。平面四角形錯体はしばしば配位不飽和であるため、5配位付加体を形成しやすく、この付加体は容易に移動挿入される。[5]ほとんどの場合、平面四角形錯体への移動経路が優先されるが、求核経路とは異なり、過剰なCOによって阻害される。[6]

反応速度への影響
- 立体効果ひずみ – 平面四角形錯体のキレート骨格の立体ひずみが増加すると、カルボニル基とメチル基が互いに近づき、挿入反応の反応性が増加します。[6]
- 酸化状態– 金属の酸化は挿入反応速度を上昇させる傾向がある。この機構における主な律速段階は、メチル基がカルボニル配位子へ移動し、アセチル炭素により多くの部分正電荷を与えることで金属を酸化し、反応速度を上昇させることである。[7]
- ルイス酸– ルイス酸も反応速度を上昇させます。これは金属の酸化が炭素の正電荷を増加させるのと同様の理由によるものです。ルイス酸はCOの酸素と結合して電荷を除去し、炭素の求電子性を高めます。これにより反応速度は最大10 8倍まで上昇し、形成される錯体は十分に安定しているため、金属に結合するCOがなくても反応は進行します。[7]
- 脱離基の電気陰性度- 脱離アルキル基の電気陰性度が増加すると、金属-炭素結合相互作用が安定化し、移動に必要な活性化エネルギーが増加し、反応速度が低下します。[8]
- トランス効果– 八面体または平面四角形の錯体中の配位子は、それがトランスである基の反応性に影響を与えることが知られています。 この配位子の影響はしばしばトランス影響と呼ばれ、その強さは配位子間で異なります。トランス影響のある配位子の一部は、トランス効果の最も高いものから最も低いものの順で次のとおりです。 [5] アリール、アルキル > NR 3 > PR 3 > AsR 3 > CO > Cl 。トランス影響が大きい配位子ほど、活性部位に大きい求電子性を付与します。 CO 基の求電子性を高めると反応速度が大幅に上昇することが実験的に示されていますが、メチル基の求電子性を低下させると反応速度がわずかに上昇します。 これは、平面四角形の [(PN)M(CO)(CH 3 )] 錯体と CO (PN は二座リンまたは窒素結合配位子) を反応させることで実証できます。この反応は、メチル基がトランス-PでCOがトランス-Nの場合、より電気陰性度の高い窒素のトランス影響が大きいため、はるかに高い収率で進行します。 [6]
逆反応
CO 挿入の逆であるアルデヒドの脱炭酸反応は、よく知られた反応です。
- RCHO → RH + CO
この反応が広く実施されていない理由の一つは、アルカンがアルデヒドの前駆体よりも有用性が低いためです。さらに、押し出されたCOの解離が遅いため、この反応は触媒的に行われることはほとんどありません。[9]有機アルデヒドからのCOの押し出しは、ウィルキンソン触媒を用いた最も有名な実証例です。[10]
- RhCl(PPh 3 ) 3 + RCHO → RhCl(CO)(PPh 3 ) 2 + RH + PPh 3
合成におけるこの基本的な有機金属反応の例については、 辻・ウィルキンソン脱炭酸反応を参照してください。
他の酸化物の挿入
多くの求電子性酸化物は、二酸化硫黄、二酸化炭素、一酸化窒素などの金属炭素結合に挿入されます。これらの反応は実用的意義が限られているか全くありませんが、歴史的に興味深いものです。遷移金属アルキルの場合、これらの酸化物は求電子剤として振る舞い、金属と比較的求核的なアルキル配位子との間の結合に挿入されます。金属二酸化硫黄錯体に関する記事で議論されているように、SO 2の挿入は特に詳細に研究されています。SO 2は挿入され、金属中心に応じてO-スルフィネートとS-スルフィネートの両方を与えます。 [11]平面四角形のアルキル錯体の場合、付加物の形成を含む前平衡が想定されます。[12]
金属-炭素結合へのアルケンの挿入
アルケンの金属-炭素間挿入は重要です。エチレンとプロピレンのチタンアルキルへの挿入は、ポリエチレンとポリプロピレンの主な原料であるチーグラー・ナッタ触媒の基礎です。この技術の大部分は不均一触媒を用いていますが、均一系における原理と観察は固体触媒にも適用できると広く考えられています。関連技術としては、洗剤の前駆体を生産するシェル高級オレフィンプロセスがあります。

アルケン重合の段階。段階iはモノマーと金属の結合であり、段階iiは移動挿入段階である。これらの段階は金属中心の片側から反対側へと交互に進行し、各ポリマー鎖について何度も繰り返される。四角で囲まれた部分は、空位(または極めて不安定な)配位部位を表す。

機構
オレフィン挿入速度に影響を与える要因には、金属とオレフィン炭素間の結合形成の初期段階を含む、環状平面四中心遷移状態の形成が含まれる。この遷移状態から、β炭素に部分的な正電荷が形成され、最初に金属に結合した炭素に部分的な負電荷が形成されることがわかる。この分極は、その後に観察される負に帯電した炭素/水素と正に帯電したβ炭素間の結合の形成、および金属-α炭素結合の同時形成を説明する。この遷移状態はまた、オレフィン挿入反応速度に最も強く寄与する2つの要因、すなわち(i)最初に金属に結合したアルキル基の軌道の重なり、および(ii)金属-アルキル結合の強度を浮き彫りにする。部分的に正のβ炭素と部分的に負の水素/アルキル基炭素間の軌道の重なりが大きいほど、新しいCC結合の形成が促進される。金属-アルキル結合の強度が増すにつれて、金属と水素/アルキル炭素結合の間の結合が切断され、α-炭素とβ-炭素との2つの新しい結合が形成される速度が遅くなり、挿入反応の速度が低下します。[13]
M–H結合へのアルケンの挿入
アルケンの金属-水素結合への挿入は、水素化反応およびヒドロホルミル化反応における重要なステップです。この反応では、アルケンとヒドリド配位子が触媒の配位圏内で結合します。水素化反応では、生成したアルキル配位子が別のヒドリドと結合してアルカンを生成します。アルキンの水素化にも同様の反応が当てはまります。アルケニル配位子がヒドリドと結合してアルケンが脱離します。

機構
機構の観点から見ると、アルケンのM–H結合への挿入とM–C結合への挿入は同様に説明されます。どちらも、置換度の低い炭素を金属側に配置する四員環遷移状態を伴います。
金属-水素結合へのオレフィン挿入の逆反応はβ-水素化物脱離である。微視的可逆性の原理によれば、β-水素化物脱離の機構は、金属水素化物結合へのアルケンの挿入と同じ経路を辿る。β-水素化物脱離の第一条件は、金属に対してβ位に水素が存在することである。β-脱離には、引き抜かれる水素を収容できる金属上の空配位位置が必要である。[14]
産業用途
カルボニル化
カルボニル基の移動挿入の広く利用されている2つの用途は、ヒドロホルミル化とメタノールのカルボニル化による酢酸の製造である。前者は、アルケン、水素、一酸化炭素をアルデヒドに変換する。カルボニル化による酢酸の製造は、2つの類似した工業プロセスを経て進行する。より伝統的なのはモンサント酢酸プロセスであり、これはロジウム-ヨウ素触媒を使用してメタノールを酢酸に変換する。このプロセスは、関連するイリジウム触媒[Ir(CO) 2 I 2 ] − ( 1 )を使用するカティバプロセスに取って代わられた。[15] [16] 2002年までに、酢酸の世界の年間生産量は600万トンに達し、そのうち約60%がカティバプロセスによって生産された。[15]
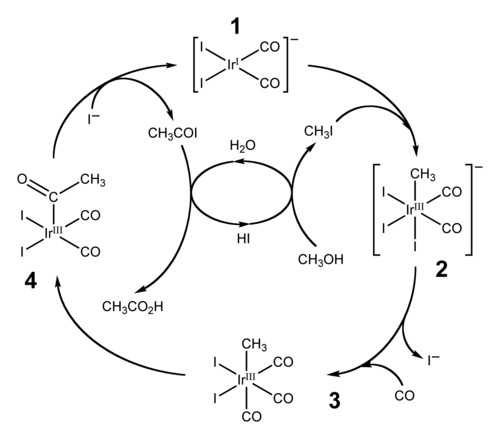
Cativaプロセスの触媒サイクル

上に示したCativaプロセスの触媒サイクルには、挿入段階と脱挿入段階の両方が含まれます。ヨウ化メチルと( 1 )の酸化付加反応は、イリジウム(I)中心の炭素-ヨウ素結合への形式的挿入を伴い、一方、( 3 )から( 4 )への反応は、イリジウム-炭素結合への一酸化炭素の移動的挿入の例です。活性触媒種は、 ( 4 )からのヨウ化アセチルの還元的脱離、すなわち脱挿入反応によって再生されます。 [15]
アルケン重合
アルケン挿入反応の工業的応用としては、金属触媒を用いたポリエチレンおよびポリプロピレンの合成が挙げられる。これらの変換反応は、典型的には、アルミニウムアルキルで活性化された三塩化チタンを触媒として不均一系で行われる。この技術はチーグラー・ナッタ触媒として知られている。[17]これらの反応では、エチレンが金属チタンに配位し、その後アルケンが挿入される。これらのステップは複数回繰り返すことができ、高分子量ポリマーの生成につながる可能性がある。
参考文献
- ^ エルシェンブロイヒ、C. (2006)。有機金属。ワインハイム: ワイリー-VCH。ISBN 978-3-527-29390-2。
- ^ Hartwig, JF (2010).有機遷移金属化学 結合から触媒まで. ニューヨーク: University Science Books. ISBN 978-1-891389-53-5。
- ^ Yadav, MS (2005). 無機化学クイックレビュー. Anmol Publications. p. 244. ISBN 978-81-261-1898-4。
- ^ F. Calderazzo, FA Cotton. 「メチルマンガンペンタカルボニルのカルボニル化とアセチルマンガンペンタカルボニルの脱カルボニル化」Inorg. Chem. , 1962, 1, 30–36. https://doi.org/10.1021/ic50001a008.
- ^ abc Anderson, GK; Cross, RJ (1984). 「四角平面錯体のカルボニル挿入反応」. Acc. Chem. Res. 17 (2): 67– 74. doi :10.1021/ar00098a005.
- ^ abc Cavell, KJ (1996). 「金属-炭素結合への移動挿入に関する最近の基礎研究」. Coord. Chem. Rev. 155 (11): 209– 243. doi :10.1016/S0010-8545(96)90182-4.
- ^ ab Alexander, JJ (1985). 「遷移金属-炭素結合への挿入」 Hartley; Patai (編).金属-炭素結合の化学第2巻. John Wiley & Sons. pp. 339– 400. doi :10.1002/9780470771747.ch5. ISBN 978-0-470-77174-7。
- ^ Shusterman, AJ; Tamir, I.; Pross, A. (1988). 「有機金属転移反応のメカニズム.配置混合(CM)アプローチ」. J. Organomet. Chem. 340 (2): 203– 222. doi :10.1016/0022-328X(88)80076-7.
- ^ Fristrup, Peter; Kreis, Michael; Palmelund, Anders; Norrby, Per-Ola; Madsen, Robert (2008). 「ロジウム触媒によるアルデヒドの脱炭酸反応の機構:実験と理論の複合研究」. J. Am. Chem. Soc. 130 (15): 5206– 5215. Bibcode :2008JAChS.130.5206F. doi :10.1021/ja710270j. PMID 18303836. S2CID 207119793.
- ^ Ohno, K.; Tsuji, J. (1968). 「貴金属化合物による有機合成. XXXV. ロジウム錯体を用いたアルデヒドおよびアシルハライドの新規脱カルボニル化反応」. J. Am. Chem. Soc. 90 (1): 99– 107. Bibcode :1968JAChS..90...99O. doi :10.1021/ja01003a018.
- ^ ダグラス、マクダニエル、アレクサンダー (1994).無機化学の概念とモデル(第3版). John Wiley & Sons, Inc. ISBN 978-0-471-62978-8。
- ^ Puddephatt, RA; Stalteri, MA (1980). 「メチル遷移金属結合またはフェニル遷移金属結合への二酸化硫黄の挿入の競合」. Journal of Organometallic Chemistry . 193 : C27 – C29 . doi :10.1016/S0022-328X(00)86091-X.
- ^ Burger, BJ; Thompson, ME; Cotter, WD; Bercaw, JE (1990). 「ペルメチルスキャンドセンアルキル錯体におけるエチレン挿入とβ水素脱離.エチレンのチーグラー・ナッタ重合における連鎖成長と停止段階の研究」. J. Am. Chem. Soc. 112 (4): 1566– 1577. Bibcode :1990JAChS.112.1566B. doi :10.1021/ja00160a041.
- ^ Crabtree, RH (2009).遷移金属の有機金属化学. John Wiley and Sons . p. 192. ISBN 978-0-470-25762-3。
- ^ abc Jones, Jane H. (2000). 「酢酸製造のためのCativaプロセス」. Platinum Metals Review . 44 (3): 94– 105. doi : 10.1595/003214000X44394105 .
- ^ Sunley, GJ; Watson, DJ (2000). 「イリジウムを用いた高生産性メタノールカルボニル化触媒 - 酢酸製造のためのCativaプロセス」. Catalysis Today . 58 (4): 293– 307. doi :10.1016/S0920-5861(00)00263-7.
- ^ Kissin, YV (2008). 「アルケン重合触媒系における遷移金属成分および助触媒の合成、化学組成、構造」.遷移金属触媒によるアルケン重合反応. アムステルダム: Elsevier . pp. 207– 290. ISBN 978-0-444-53215-2。
外部リンク
- 渡り鳥の挿入
- 有機金属ハイパーテキストブック:挿入反応
- 移動挿入